

Enhancing Common Technical Document (CTD) Quality Modules: Practical Insights and Recommendations
Developing a comprehensive Common Technical Document (CTD) is crucial for drug applications, encompassing investigational and marketing registration phases. While guidance documents outline the technical information and its organization within CTD Module 3, sponsors retain flexibility in presenting data and formatting important messages within their application. The key to expediting the review and approval process by regulatory authorities, such as the FDA, is preparing a high-quality and well-designed Module 3 CTD. This article provides practical insights and recommendations to optimize the potential for a successful outcome.
CTD Structure and Content for CMC Submission
Over the last 25 years, the International Conference on Harmonization (ICH) has made significant strides in unifying drug application dossiers. The resulting CTD has become the preferred format across ICH regions, including the United States. That said, it is essential to note that the CTD does not replace or supersede the regulations defined in the US Code of Federal Regulations.
The CTD serves as an agreed-upon format for presenting summaries, reports, and data, while the content must adhere to regulatory requirements and recommendations in FDA guidance documents. Furthermore, specific components may be required by individual ICH regions, necessitating the tailoring of CTD submissions to meet unique regulatory standards.
The organization of technical information within the Quality Modules of the CTD is modular and outlined in guidance documents. Nonetheless, sponsors are free to present data and format important messages as they see fit within their CTD application. Ultimately, ensuring the timeliness of FDA review and approval status for a drug’s Quality section necessitates the preparation of well-designed modules.
In the forthcoming sections, while acknowledging the inherent imbalance, I will focus on selected aspects of the Quality modules that significantly influence an application’s overall success or failure. This discussion will delve into the intricacies of producing approvable Quality Modules—a complex and technical exercise.
Critical Strategies for Maximizing Module 3 Acceptance in CTD Applications
To significantly enhance the likelihood of receiving favorable feedback on Module 3 within a CTD application, adopting a fundamental approach that prioritizes clarity, transparency, and honest discussions of both positive and negative findings is crucial. It is vital to avoid making unsubstantiated claims and to remember that undocumented information holds no weight. I emphasize the significance of employing these strategies, which can substantially impact the perception of the application.
The organization of the Module 3 section, following the CTD format, for both investigational and marketing applications is not inherently more challenging than a standard New Drug Application (NDA). Various guidance documents provide valuable assistance throughout the process. However, the “art” of presentation and scientific content can offer significant advantages in specific areas. These highlighted sections are the focus of this article series. It’s important to note that the recommendations provided are based on this author’s experience rather than existing literature.
When reviewers find an application’s content and presentation favorable, with a clear trail from statements to supporting documentation, it dramatically increases the chances of swift approval. Furthermore, a well-crafted CTD application offers an additional benefit: FDA requirements for supplemental data, typically imposed during the review process, may be transformed into postmarket obligations rather than preapproval prerequisites. However, if the reviewers are not comfortable, review issues are more likely to lead to new preapproval data requirements that could delay approval.
By employing the strategies outlined herein, sponsors can significantly improve the reception of Module 3 within their CTD applications, leading to a higher likelihood of rapid approval and minimizing additional preapproval obligations imposed during the review process.
The Structure and Significance of CTD Quality Modules
The format of the Common Technical Document (CTD) Quality Modules follows a well-defined outline, as outlined in the Table that follows. Each section within the modules has explicit requirements for content and formatting. The application consists of summary and overview discussions of chemistry, manufacturing, and controls (CMC, quality) data in Module 2, along with detailed technical data and study reports in Module 3. While the content of these modules is generally well-defined based on relevant guidance documents, there is significant flexibility and encouragement for assimilating, discussing, comparing, and contrasting data, particularly within Module 2. The length of Quality Modules can vary based on available data, providing opportunities for creativity, storytelling, and crafting cohesive arguments to help regulatory bodies understand the product. The modular format, with different levels of detail, enables the presentation of an overall picture while making all supporting information available. Given the more significant number of modular components within the Quality Module and the complexity of the data, it is essential to cross-reference sections within and between modules carefully.
While each module of the CTD Quality Module serves a specific function, the sections for creative and informative content primarily lie in select conformance discussions. These sections allow for the integration of data from development studies, presenting both the strengths and limitations of the data. They allow FDA reviewers to comprehend the broader context at various levels of detail. Clear and compelling presentations in these modules are crucial for the application’s success.
Regulatory bodies in the United States, Canada, European Union, and Japan require the CTD in the specified content and format for IND and NDA (or equivalent) approval. This agreement, led by ICH Harmonization, streamlines the application process for seeking regulatory approvals. The CTD’s groundwork and preparation are critical as they are the foundation for regulatory filings. While the FDA does not conduct site audits during the investigational stage, Module 3 of the CTD should focus on Regulatory CMC documentation related to substance and product quality. Providing accurate and detailed information in this section reduces delays and costs, adding significant value to the process.
When preparing the CMC section of the CTD, it is vital to provide a comprehensive interpretation and analysis of regulations and requirements to support the proposed strategy. The focus of Module 3 is to demonstrate the “quality” of the candidate intermediate, drug substance, or drug product to satisfy regulatory authorities. Ensuring accuracy and completeness in this section saves valuable time and effort since the CTD document is accepted by regulatory bodies in major pharmaceutical markets worldwide.
While every section of CTD Module 3 plays a crucial role in supporting the ultimate approval of a new drug, certain areas now hold particular significance. Traditionally, the manufacturing process description (S.2.2 Description of Manufacturing and P.3.2 Batch Formula), drug substance and drug product specifications (S.4.1 Specifications and P.5.1), analytical procedures (S.4.2 and P.5.2), and stability (S.7 and P.8) sections served as an introduction and summary of all compliance data available for the drug. They continue to receive a thorough review.
Now more than ever, conformance sections (S.2.6 Manufacturing Process Development, S.4.5 Justification of Specifications, P.2 Pharmaceutical Development, P.5.5 JOS, etc.) provide an opportunity to craft discussions, arguments, explanations, and justifications, highlighting supportive data while putting less-than-stellar findings into perspective. These sections can significantly influence reviewers’ views, making it preferable to proactively address potential challenges within these sections rather than waiting for regulatory reviewers to notice problematic data.
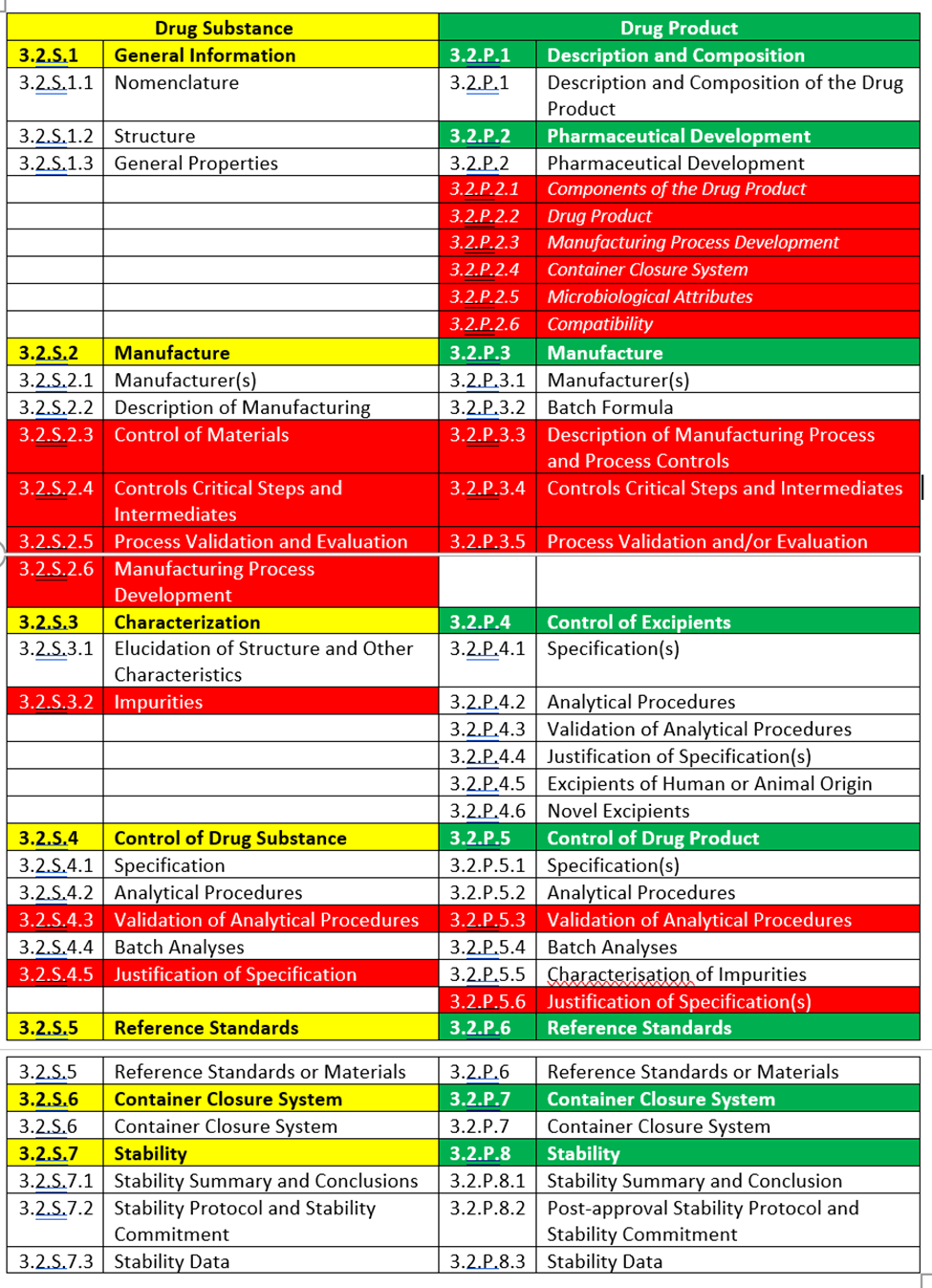
Common Technical Document (CTD) Module 3
Module 2, the Quality Overall Summary (QOS)
Module 2 QOS, plays a critical role in drug development. While it is ideal for writing Module 2 after completing all subsections of Module 3, the realities of modern drug development often make this difficult. Therefore, one of the initial and critical steps for sponsors is to assemble a cross-disciplinary team with the necessary technical expertise and collaborative skills. Building an effective team, guided by strong leadership and clear goals, significantly enhances the chances of success. By focusing on primary marketing objectives, addressing weaknesses in the data package with scientifically driven strategies, and defining immediate and long-term outcomes, the team can ensure that the final Module 2 documents align with the intended goals.
CTD Module 2 serves a similar purpose to the application summary in the traditional New Drug Application (NDA) format, providing an overview of Module 3. Building Module 2 is an iterative process, and efficiencies can be achieved by leveraging pre-NDA meeting documents as the basis for eventual Module 2 summaries. Developing a clear and consistent message during pre-NDA efforts maximizes the efficiency of Module 2 preparations.
The QOS, or CTD Module 2.3, is a presentation that should provide the quality reviewer and other reviewers with a comprehensive overview of Module 3. The QOS should follow the general format and outline of the detailed data in Module 3, emphasizing critical vital parameters of the product. It should summarize information on potential and actual impurities from the active ingredient’s synthesis, manufacturing, or degradation. The basis for setting acceptance criteria for individual and total impurities should be summarized, along with the qualification of proposed impurity limits. Module 2.3 should also outline impurity levels in batches of the drug substance used in nonclinical studies, clinical trials, and typical batches manufactured by the proposed commercial process. Explanations and justifications can be included in Module 2.3, such as providing a rationale for deviating from guidance. Much of the requested information, including tables, figures, and process diagrams, can be imported directly from Module 3. Other details, like a brief overview of the manufacturing process and a summary of significant manufacturing changes throughout development, should be presented as highlights to ensure all reviewers gain a basic understanding of the product, not just those specialized in CMC.
The QOS should include discussions of critical issues that warrant integrating information from manufacturing, clinical, and nonclinical programs. Examples of such integrated cases are the qualification of impurities through toxicologic studies and human risk assessment based on safety evaluations during clinical trials. It could also explain the need for special reprocessing steps, such as minimizing toxic contaminants or optimizing processes with expensive ingredients or poor yields.
The QOS separately covers the drug substance (active ingredient) and drug product (dosage form). For both entities, the QOS aims to convey critical concepts such as characterization, consistency (batch to batch), process control, comparability throughout development (summarizing Module 3 reports), and establishing the connection between clinical drug supplies and the proposed marketed product.
Tabular presentations are preferred to compare and contrast data across batches, time, and manufacturing process improvements whenever possible. Comparative exhibits effectively highlight manufacturing consistencies and changes to enhance product efficacy and toxicity profile or scale up production from nonclinical and early clinical testing to significant efficacy protocols. The QOS should generally be at most 25 – 30 pages of text, excluding tables and figures. However, the document could be longer for biotech products and those manufactured using complex processes. However, it should still be presented as highlights to ensure all reviewers can grasp the basic understanding of the product.
Module 3: The Quality Module
Module 3 encompasses the drug substance (active ingredient) and drug product sections, each providing introductory presentations of technical drug information, processes, key parameters, and justifications supported by qualification and validation studies. These reports serve as detailed evidence that a drug’s characteristics are well-defined and controlled, ensuring that each new lot produced is identical to the previous one. Demonstrating control and reproducibility in drug manufacturing is crucial for FDA reviewers to consider approving a new drug. In the case of an older drug, much of the manufacturing information can be cross-referenced to an existing Drug Master File from the active ingredient manufacturer, minimizing repetition in Module 3.
It is impractical to summarize all the CMC sections that constitute Module 3. However, certain areas deserve particular attention. Section 3.2.R, titled “Regional Information,” includes additional drug substance and product details specific to different regulatory authorities. Applicants are encouraged to stay updated on evolving requirements and actively engage in dialogue with these authorities to address country-specific needs for their products.
Module 3 and the associated development work should go beyond presenting isolated data points and instead tell a cohesive story. The manufacturing development program undergoes significant evolution for most drugs, resulting in substantial differences between the early-stage drug substance or product and the proposed marketing version. Describing these manufacturing development changes effectively requires convincing FDA reviewers to consider and integrate nonclinical and clinical data collected at various points during development, even if the drugs differ significantly at those stages.
One challenge in creating a coherent Module 3 arises because CMC data often originates from multiple sources. While some organizations handle all chemistry development in-house, relying on contributions from both in-house and external parties is more common. As drug development accelerates, the pressure to generate batches of drug substance and drug product for nonclinical and clinical trials intensifies. Good Manufacturing Practices (GMP) standards necessitate rigorous documentation for analytical and stability programs supporting manufacturing. Simultaneously, manufacturing experts explore more efficient process schemes and frequently consider alternate contractors to reduce costs and avoid reliance on a single source. All these changes demand thorough documentation and evidence of control, ideally beginning from the project’s initiation and planning proactively as far ahead as possible. It is crucial to start Module 3 on the right footing, as attempting to reconstruct information from primary sources after the fact, especially if the responsible individuals are no longer available or links are missing, can be exceptionally challenging.
Another distinctive aspect of Module 3 is the requirement for development reports. These reports encompass drug substance, drug product, and analytical aspects of pharmaceutical development. They must effectively document the historical evolution of these three components throughout the product’s development lifecycle. FDA reviewers need a clear understanding of how the drug has evolved and ultimately agree that all nonclinical and clinical data gathered during development provide informative and relevant insights into the eventual marketed drug product. Given that drugs rarely remain unchanged during years of development, it is essential to document all ongoing chemistry and manufacturing changes and comprehend the impact of product differences. Development reports may require input from chemists and professionals with expertise in effective communication. Poorly prepared or unconvincing development reports can lead to a cycle of FDA queries and sponsor responses, significantly prolonging the review process and delaying approval times.
Conclusion
In conclusion, the successful preparation and presentation of Module 3 following the Common Technical Document (CTD) format significantly contribute to the chances of a favorable review and approval by regulatory authorities. The key to maximizing the likelihood of approval lies in pursuing clarity, avoiding exaggerations, and openly addressing any negative findings or deficiencies. It is crucial to substantiate claims and refrain from undocumented assertions, as unsupported statements hold little weight in the evaluation process.
While organizing Module 3 according to the CTD format is not inherently more challenging than a standard New Drug Application (NDA), certain sections offer opportunities to leverage both scientific rigor and the art of presentation. Although derived from the author’s experience rather than existing literature, the recommendations provided in this article highlight the sections where additional attention can give you significant advantages.
When reviewers can readily comprehend the content and follow the logical flow from statements to supporting documentation, the chances of expedited approval are greatly enhanced. Moreover, a well-crafted CTD application increases the likelihood of approval without additional data requirements imposed during the review. It reduces the risk of preapproval obligations, as review issues are more likely to result in post market conditions. A high-quality CTD application fosters a favorable impression among reviewers, facilitating a smoother and faster approval process.

